
..Redacción.
El Comité de Medicamentos de Uso Humano (CHMP) de la Agencia Europea de Medicamentos (EMA) ha concluido el arbitraje realizado por la presencia de nitrosaminas en los medicamentos del grupo de los “sartanes” incluyendo candesartán, irbesartán, losartán, olmesartán y valsartán. Los laboratorios fabricantes de principios activos incluidos en el arbitraje deberán revisar sus procesos de fabricación para evitar la presencia de nitrosaminas y contarán con un plazo de dos años para hacerlo.
La revisión por parte del CHMP ha afectado a los medicamentos que tienen como principio activo candesartán, irbesartán, losartán, olmesartán y valsartán, todos ellos antagonistas de los receptores de la angiotensina II
Por su parte, la Agencia Española de Medicamentos y Productos Sanitarios (AEMPS) ha ido informando desde julio del año pasado sobre la retirada del mercado estos fármacos ante la presencia de impurezas que previamente no habían sido detectadas.

 La revisión por parte del CHMP ha afectado a los medicamentos que tienen como principio activo candesartán, irbesartán, losartán, olmesartán y valsartán, todos ellos antagonistas de los receptores de la angiotensina II. Indicados para tratar la hipertensión arterial y enfermedades cardíacas o renales, actúan bloqueando la acción de la angiotensina II, una hormona que contrae los vasos sanguíneos y hace que aumente la presión arterial.
La revisión por parte del CHMP ha afectado a los medicamentos que tienen como principio activo candesartán, irbesartán, losartán, olmesartán y valsartán, todos ellos antagonistas de los receptores de la angiotensina II. Indicados para tratar la hipertensión arterial y enfermedades cardíacas o renales, actúan bloqueando la acción de la angiotensina II, una hormona que contrae los vasos sanguíneos y hace que aumente la presión arterial.
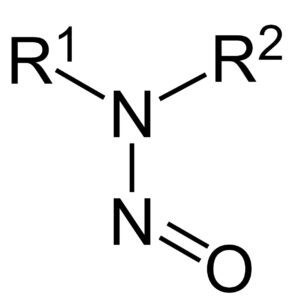
Estas impurezas se pueden formar durante la fabricación de medicamentos que contienen un anillo específico en su estructura, conocido como anillo tetrazol
Impurezas detectadas
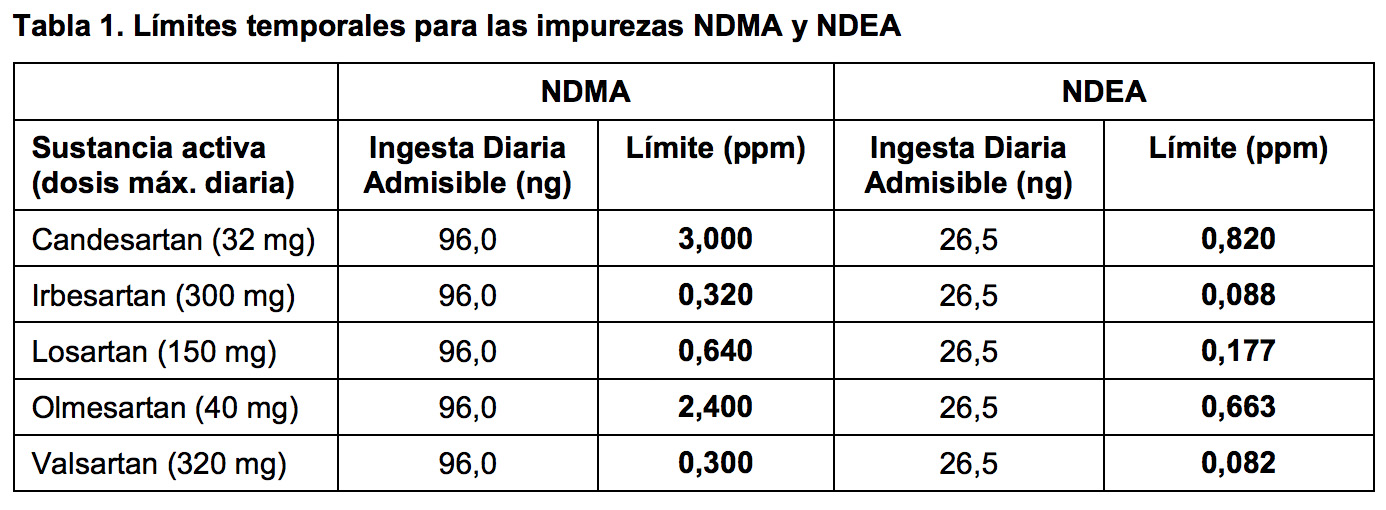
Las impurezas detectadas son N-Nitrosodimetilamina (NDMA) y NNitrosodietilamina (NDEA) y previamente no estaban identificadas en este grupo de medicamentos y, por lo tanto, no se detectaban en los ensayos de rutina. Sin embargo, están clasificadas como probablemente carcinógenas en humanos.
La revisión del CHMP comenzó el 5 de julio de 2018 solo para valsartán, pero el 20 de septiembre de 2018 se extendió para incluir el resto de sartanes ya que estas impurezas se pueden formar durante la fabricación de aquellos medicamentos que contienen un anillo específico en su estructura, conocido como anillo tetrazol bajo determinadas condiciones (disolventes, reactivos y otros materiales de partida). Además, también es posible que las impurezas estuvieran presentes en ciertos sartanes por la utilización durante el proceso de fabricación de equipos o reactivos contaminados.
Los fabricantes de sartanes deberán revisar sus procesos de fabricación y establecer ensayos que permitan detectar cantidades mínimas de estas nitrosaminas
 Revisión de los procesos de fabricación
Revisión de los procesos de fabricación
Ahora, los fabricantes de sartanes deberán revisar sus procesos de fabricación y establecer ensayos que permitan detectar cantidades mínimas de estas nitrosaminas. Para realizar los cambios necesarios en sus procesos de fabricación, las compañías contarán con un periodo de transición de dos años durante el cual se ha establecido temporalmente límites estrictos para estas impurezas en línea con las guías actuales.
Estos límites se basan en la ingesta diaria admitida para cada una de las impurezas, derivados de estudios en animales: 96,0 nanogramos para NDMA y 26,5 nanogramos para NDEA. Cuando se dividen estos niveles entre la dosis máxima diaria para cada una de las sustancias activas estudiadas se obtienen los límites en partes por millón. Tras este periodo, los fabricantes deberán demostrar que los medicamentos no tienen niveles cuantificables de estas impurezas, descartando aquellos productos que contengan incluso niveles menores de NDEA o NDMA evitando su uso en la UE.
Las recomendaciones realizadas por la EMA sobre las impurezas NDMA y NDEA se enviarán a la Comisión Europea que será quien tome la decisión formal
La EMA y las autoridades nacionales van a continuar investigando la presencia de nitrosaminas en los medicamentos, incluyendo otras impurezas como la N-nitrosoetilisopropilamina (EIPNA), la N-nitrosodiisopropilamina (DIPNA) y el ácido N-nitroso-N-metilaminobutírico (NMBA). También considerarán lo aprendido tras esta revisión para mejorar la identificación y tratamiento de las impurezas en los medicamentos. Por último, las recomendaciones realizadas por la EMA sobre las impurezas NDMA y NDEA se enviarán a la Comisión Europea que será quien tome la decisión formal.






